1.2 Using NCBI-BLAST
This book begins by exploring the BLAST pages on the NCBI web site. The NCBI, part of the National Institutes of Health, is a U.S. government-funded center for the curation and presentation of public biological knowledge. The NCBI is a public repository for DNA and protein sequences (GenBank), but it's far more than just a data storehouse. The NCBI also maintains a comprehensive medical publication archive (PubMed), distributes many tools for biological analyses (NCBI toolbox), and puts together its own tools for making the most use of the data that it stores (LocusLink, UniGene, RefSeq, Taxonomy browser). Most importantly, for our purposes, it's where the BLAST algorithm was first developed (Altschul et al., 1990) and where it can be obtained, distributed, and used for free without restrictions. Anyone with access to the Internet can run a BLAST search and explore the plethora of genetic resources that have been amassed and curated by the NCBI over the years.
You'll get the most out of this chapter if you follow along with a web browser. Begin by going to the BLAST homepage at http://www.ncbi.nlm.nih.gov/BLAST.
1.2.1 Choosing the BLAST Program
Without explaining all of the options presented on the homepage, let's get right into it with a default BLASTN search. Choose "Standard nucleotide-nucleotide BLAST [blastn]" as shown in Figure 1-1. BLASTN is a program that compares a nucleotide query sequence to a database of nucleotide sequences.
Figure 1-1. NCBI BLAST home page

1.2.2 Entering the Query Sequence
After choosing the kind of search you want to perform, the next step is to define the sequence with which to search. There are three options for this: paste in the bare sequence, paste in a file in FASTA format, or enter a valid NCBI identifier. You can just start typing a sequence in the search box; however, when the search is done, there will be no identifier to describe the sequence you entered. After several such searches, the lack of an identifier will make it difficult to keep track of which results go with which sequence. The second option allows you to define the sequence using the FASTA format. The FASTA format is described in detail in Chapter 11, but the basic specifications are that it's a text file beginning with a greater than sign (>) followed by an identifier and a definition line, which is then proceeded by the one-letter nucleotide or peptide sequence on subsequent lines. Let's use the following sequence:
>gi|11611818|gb|AF287139.1|AF287139 Latimeria chalumnae Hoxa-11 gene, partial cds TACTTGCCAAGTTGCACCTACTACGTTTCGGGTCCCGATTTCTCCAGCCTCCCTTCTTTTTTGCCCCAGACCCCGTCTTCTCG CCCCATGACATACTCCTATTCGTCTAATCTACCCCAAGTTCAACCTGTGAGAGAAGTTACCTTCAGGGACTATGCCATTGATA CATCCAATAAATGGCATCCCAGAAGCAATTTACCCCATTGCTACTCAACAGAGGAGATTCTGCACAGGGACTGCCTAGCAACC ACCACCGCTTCAAGCATAGGAGAAATCTTTGGGAAAGGCAACGCTAACGTCTACCATCCTGGCTCCAGCACCTCTTCTAATTT CTATAACACAGTGGGTAGAAACGGGGTCCTACCGCAAGCCTTTGACCAGTTTTTCGAGACGGCTTATGGCACAACAGAAAACC ACTCTTCTGACTACTCTGCAGACAAGAATTCCGACAAAATACCTTCGGCAGCAACTTCAAGGTCGGAGACTTGCAGGGAGACA GACGAGAAGGAGAGACGGGAAGAAAGCAGTAGCCCAGAGTCTTCTTCCGGCAACAATGAGGAGAAATCAAGCAGTTCCAGTGG TCAACGTACAAGGAAGAAGAGGTGC
Before you try to type all this into the search text box, let's look at identifiers, which are an easier and more reliable way to enter queries. The previous example of the coelacanth (Latimeria chalumnae) Hoxa-11 gene has three valid NCBI identifiers that can be entered into the search box. The three identifiers are separated by pipes (|) and designate the GI (11611818), the accession number and version (AF287139.1), and the locus (AF287139). These identifiers are explained in detail in Chapter 11. For the current search (Figure 1-2), use the locus identifier, AF287139.
Figure 1-2. Entering the query sequence
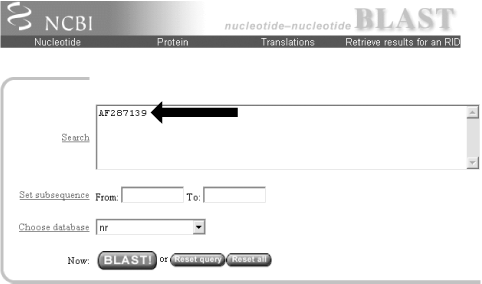
Using the locus, BLAST pulls out the FASTA file from the NCBI databases and uses it in the search just as if you had entered it all in the search box. If you are dealing with public sequence, this is the fastest and most reliable way to enter the query.
1.2.3 Choosing the Database to Search
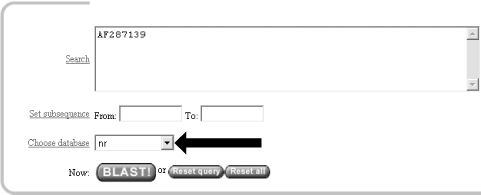
For this search, we'll leave the default database as nr (Figure 1-3). Historically, the database was curated to contain a nonredundant set of nucleotide sequences (hence nr); however, it's no longer screened to be nonredundant. Because of its comprehensive nature, nr is usually a good first start when trying to identify a novel sequence or when determining if related sequences have been described previously. The database is curated by the NCBI and consists of nucleotide sequences from all of GenBank, RefSeq, EMBL, and DDBJ. You don't need to be concerned about the details of these /-sequence sources now but just know that they provide a comprehensive set of sequences. As of January 2003, the nr database contained more than 1.5 million entries consisting of more than 7.5 billion nucleotides.
Figure 1-3. Choosing the database
1.2.4 Choosing the Parameters of the Search
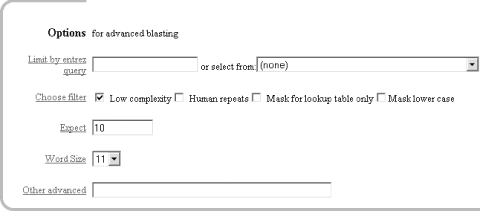
Once you enter a query sequence and choose a database, the next step is to decide on the parameters of the search (Figure 1-4). For this test case, just use the default parameters, which are low-complexity filtering, an Expect value of 10, and a word size of 11. There is also a default reward of +1 and a penalty of -3, which isn't apparent on this submission form but makes a big difference in the results you obtain. A full explanation of these parameters and how they relate to the expected results are discussed in Chapter 4, Chapter 7, and Chapter 9.
Figure 1-4. Selecting parameters
1.2.5 Choosing the Format
Once you have entered the query, selected the database, and chosen the appropriate search parameters, you must then choose the desired results format (Figure 1-5).
Figure 1-5. Choosing the format
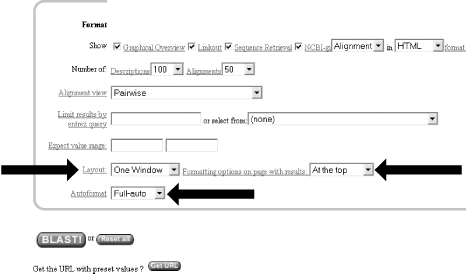
These options allow you to format the results in a number of ways. For this quick start guide, you need to change the three bottom options: "Layout," "Formatting options on page with results," and "Autoformat." "Layout" should be changed from "Two Windows" to "One Window." This keeps all the results in the current window instead of launching a separate window. The "Formatting options on page with results" should be set to "At the top." Because the NCBI has set up the BLAST pages so that the search is separate from the results, using "At the top" lets you easily explore all the different formatting options once you get your results. Now you can run the compute-intensive search once and then format it rapidly in a number of ways. The final change is to set "Autoformat" to "Full-auto." This automatically updates and formats the results page when the search is done.
1.2.6 Submitting the Search
Once you select the BLAST! button, the window changes to show the Request Identifier (RID) and the estimated time to completion (below the Format options section). The web page will update itself periodically until the search is complete (Figure 1-6).
Figure 1-6. Waiting for results
1.2.7 Viewing the Results
Once the search is complete, a results window appears. To understand all the parts of a BLAST report, break down the results window into pieces. The header of the report, shown in Figure 1-7, contains important bookkeeping information. For example, at the top is the BLAST version and date of compilation (Version 2.2.5, compiled on November 16, 2002). Also shown is the reference for the Nucleic Acids Research article, which should be used in any publication arising from using NCBI-BLAST. Following the reference is the RID, which can be copied and used to retrieve these results for up to 24 hours. Next, the query definition line and sequence length are reported along with a description of the database and its size. Also included in the header is a link to "Taxonomy reports," which shows the lineage and taxonomic breakdown of all the database matches.
Figure 1-7. Header of a BLAST report

Looking further down in the report (Figure 1-8), you can see that the body of the report begins with a graphical display of the database hits (the result of setting the Graphical Overview option) as they align to the query. At the top of the display, you can see that 72 BLAST hits passed the threshold of your search criteria (you may see more than 72 because of the rapid database growth). After the color key, the top line represents the query sequence as a solid red line with the sequence coordinates. Each line below represents one subject match with its position in relation to the query and the color-coded relative strength of the similarity. You can move your mouse over each line to see the definition line, and if you click on it, you will be taken to the actual alignment.
Figure 1-8. The body: graphical overview

The next part of the body is the summary (see Figure 1-9), which lists the one-line descriptions (set with the Descriptions option) of the database matches (also known as hits or subjects) along with the score and the E value. The hits are listed from best to worst, with high scores and low E values being better. Also included in this part, and set with the Linkout option, are links to other NCBI curated databases with more information about each hit. In this case some sequences have links in LocusLink (L) and/or UniGene (U).
Figure 1-9. The body: one-line descriptions

At the heart of the report are the actual alignments (the number of alignments displayed is controlled by the Alignments option). The definition line is listed for each subject, and then some statistics about the alignment are given (Score, Expect (E) value, Identities, and Strand), followed by the actual sequence alignment. The letters of the sequences involved in the alignment are shown with the sequence coordinates and vertical bars connecting identical letters.
Figure 1-10 shows one database match alignment from this search. The query (your input) is aligned to the subject (a chicken homeodomain-containing gene) with all high-scoring local alignments shown. Each alignment is a high-scoring segment pair (HSP) that has its own alignment statistics. There are three HSPs in this case, each with a very significant score and Expect value. Some subject sequences have an associated link "D" that allows you to download just the part of the subject that aligns with the query, plus up to 1,000 bases flanking the HSP.
Figure 1-10. The body: alignments

Finally, at the bottom of the report, after all significant alignments are shown, comes the footer containing a detailed description of the search parameters (Figure 1-11). The footer contains information about the database, including a brief description, the date posted, and the size. The footer also lists the values of the lambda, K, and H variables used in calculating E values, bit scores, and other statistics about the alignments. The significance of all these numbers are explained in detail in Chapter 4 and Chapter 7.
Figure 1-11. The footer









