5.2 The BLAST Algorithm
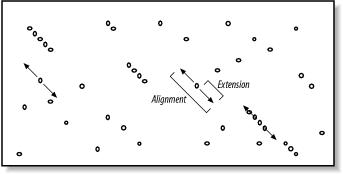
The search space between two sequences can be visualized as a graph with one sequence along the X-axis and the other along the Y-axis (Figure 5-1). Each point in this space represents a pairing of two letters, one from each sequence. Each pair of letters has a score that is determined by a scoring matrix whose values were determined empirically. (See Chapter 4 for more about scoring matrices.) An alignment is a sequence of paired letters that may contain gaps (See Chapter 3 and Chapter 4 for more about gaps). Ungapped alignments appear as diagonal lines in the search space, and the score of an ungapped alignment is simply the sum of the scores of the individual letter pairs. Alignments containing gaps appear as broken diagonals in the search space, and their score is the sum of the letter pairs minus the gap costs, which usually penalize more score points for initiating a gap than extending a gap. How can you tell the difference between two ungapped alignments and a single gapped alignment? In a BLAST report, unaligned regions aren't displayed, and gaps are represented by dashes. However, a simple change in parameters can change one into the other. The diagrams in this chapter show only one gapped alignment, which is indicated in Figure 5-1.
Figure 5-1. Search space and alignment
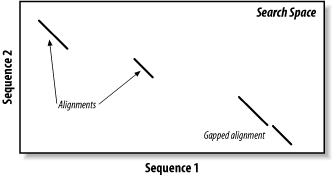
As you saw in Chapter 3, the Smith-Waterman algorithm will find the maximum scoring alignment between two sequences. Some people feel that this ability makes Smith-Waterman the gold standard of alignment algorithms, but this is true only in theory. When comparing real sequences, you may have several good alignments or none. What you really want reported is all of the statistically significant alignments; this is what BLAST does. However, unlike Smith-Waterman, BLAST doesn't explore the entire search space between two sequences. Minimizing the search space is the key to its speed but at the cost of a loss in sensitivity. You will find that the speed/sensitivity trade-off is a key concept when designing BLAST experiments. How exactly does BLAST find similarities without exploring the entire search space? It uses three layers of rules to sequentially refine potential high scoring pairs (HSPs). These heuristic layers, known as seeding, extension, and evaluation, form a stepwise refinement procedure that allows BLAST to sample the entire search space without wasting time on dissimilar regions.
5.2.1 Seeding
BLAST assumes that significant alignments have words in common. A word is simply some defined number of letters. For example, if you define a word as three letters, the sequence MGQLV has words MGQ, GQL, and QLV. When comparing two sequences, BLAST first determines the locations of all the common words, which are called word hits (Figure 5-2). Only those regions with word hits will be used as alignment seeds. This way, BLAST can ignore a lot of the search space.
Figure 5-2. Word hits
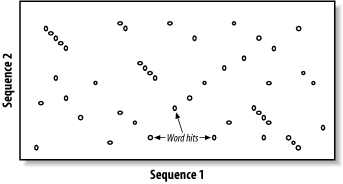
Let's take a moment to define a word hit. A simple interpretation is that a hit is two identical words. Some significant alignments don't contain any identical words, though. Therefore, BLAST employs a more useful definition of a word hit called the neighborhood. The neighborhood of a word contains the word itself and all other words whose score is at least as big as T when compared via the scoring matrix. Therefore, by adjusting T it is possible to control the size of the neighborhood, and therefore the number of word hits in the search space. Table 5-2 shows the neighborhood around the word RGD, and Example 5-1 shows a Perl script for determining the neighborhood for three-letter words.
|
BLOSUM62 |
PAM200 | ||
|---|---|---|---|
|
Word |
Score |
Word |
Score |
|
RGD |
17 |
RGD |
18 |
|
KGD |
14 |
RGE |
17 |
|
QGD |
13 |
RGN |
16 |
|
RGE |
13 |
KGD |
15 |
|
EGD |
12 |
RGQ |
15 |
|
HGD |
12 |
KGE |
14 |
|
NGD |
12 |
HGD |
13 |
|
RGN |
12 |
KGN |
13 |
|
AGD |
11 |
RAD |
13 |
|
MGD |
11 |
RGA |
13 |
|
RAD |
11 |
RGG |
13 |
|
RGQ |
11 |
RGH |
13 |
|
RGS |
11 |
RGK |
13 |
|
RND |
11 |
RGS |
13 |
|
RSD |
11 |
RGT |
13 |
|
SGD |
11 |
RSD |
13 |
|
TGD |
11 |
WGD |
13 |
Example 5-1. Determining the neighborhood for three-letter words
#!/usr/bin/perl -w
use strict;
die "usage: $0 <matrix> <word> <threshold>\n" unless @ARGV == 3;
my ($matrix_file, $WORD, $T) = @ARGV;
my @W = split(//, $WORD);
die "words must be 3 long\n" unless @W == 3;
my @A = split(//, "ARNDCQEGHILKMFPSTWYVBZX*"); # alphabet
my %S; # Scoring matrix
# Read scoring matrix - use those provided by NCBI-BLAST or WU-BLAST
open(MATRIX, $matrix_file) or die;
while (<MATRIX>) {
next unless /^[A-Z\*]/;
my @score = split;
my $letter = shift @score;
for (my $i = 0; $i < @A; $i++) {
$S{$letter}{$A[$i]} = $score[$i];
}
}
# Calculate neighborhood
my %NH;
for (my $i = 0; $i < @A; $i++) {
my $s1 = $S{$W[0]}{$A[$i]};
for (my $j = 0; $j < @A; $j++) {
my $s2 = $S{$W[1]}{$A[$j]};
for (my $k = 0; $k < @A; $k++) {
my $s3 = $S{$W[2]}{$A[$k]};
my $score = $s1 + $s2 + $s3;
my $word = "$A[$i]$A[$j]$A[$k]";
next if $word =~ /[BZX\*]/;
$NH{$word} = $score if $score >= $T;
}
}
}
# Output neighborhood
foreach my $word (sort {$NH{$b} <=> $NH{$a} or $a cmp $b} keys %NH) {
print "$word $NH{$word}\n";
}
The proper value for T depends on both the values in the scoring matrix and the balance between speed and sensitivity. Higher values of T progressively remove more word hits (see Figure 5-3) and reduce the search space. This makes BLAST run faster, but increases the chance of missing an alignment.
Figure 5-3. How T affects seeding
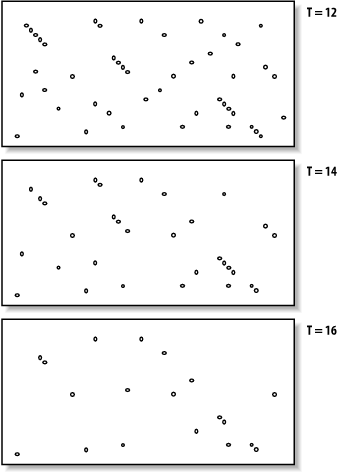
Word size (W) is another variable that controls the number of word hits. It's easy to see why a word size of 1 will produce more hits than a word size of 10. In general, if T is scaled uniformly with W, smaller word sizes increase sensitivity and decrease speed. The interplay between W, T, and the scoring matrix is critical, and choosing them wisely is the most effective way to control the speed and sensitivity of BLAST.
In Figures Figure 5-2 and Figure 5-3, you may have noticed that word hits tend to cluster along diagonals in the search space. The two-hit algorithm, as it is known, takes advantage of this property by requiring two word hits on the same diagonal within a given distance (see Figure 5-4). Smaller distances isolate more single word hits and further reduce the search space. The two-hit algorithm is an effective way to remove meaningless, neighborless word hits and improve the speed of BLAST searches.
Figure 5-4. Isolated and clustered words
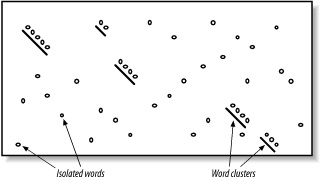
5.2.1.1 Implementation details
The descriptions thus far have been mostly theoretical. However, some implementation details are worth discussing. In NCBI-BLAST, BLASTN is very different from the other, protein-based algorithms. BLASTN seeds are always identical words; T is never used. To make BLASTN faster, you increase W and to make it more sensitive, you decrease W. The minimum word size is 7. The two-hit algorithm isn't used in BLASTN searches because word hits are generally rare with large, identical words. BLASTP and the other protein-based programs use word sizes of 2 or 3. To make protein searches faster, you set W to 3 and T to a large value like 999, which removes all potential neighborhood words. The two-hit distance is set to 40 amino acids by default, so words don't have to be clustered as closely as they appear in the figures. In principle, setting the two-hit distance to a smaller value also increases speed, but in practice, its effect is insubstantial.
In WU-BLAST, you may set W to any value for any program. If W is set to 5 or more, neighborhood word scores aren't used; they are computed only by explicitly assigning a value for T. High values of W in conjunction with moderate values of T can lead to immense memory requirements, so it is best not to set T when W is 6 or more. To alter the speed/sensitivity of WU-BLAST you can use a variety of combinations of W, and T, and you can also employ the two-hit algorithm.
The statistical model underlying BLAST assumes the letters are independent of one another so that the words MQV and MVQ have the same probability of occurring. However, certain combinations occur in biological sequences much more often than expected. These are usually low-complexity sequences?for example, FFF (see Chapter 2 and Chapter 4). Low-complexity sequences are often of little biological interest and aligning them wastes CPU cycles. Masking these regions is therefore common. Both NCBI-BLAST and WU-BLAST let you replace these regions with N's or X's, which have negative scores when aligned with a nucleotide or amino acid. A more useful technique, termed soft masking, prevents seeding in such regions, but lets alignments extend through them.
5.2.2 Extension
Once the search space is seeded, alignments can be generated from the individual seeds. We've drawn the alignment as arrows extending in both directions from the seeds (Figure 5-5), and you'll see why this is appropriate. In the Smith-Waterman algorithm (Chapter 3), the endpoints of the best alignment are determined only after the entire search space is evaluated. However, because BLAST searches only a subset of the space, it must have a mechanism to know when to stop the extension procedure.
Figure 5-5. Generating alignments by extending seeds
To make this clearer, we'll try aligning two sentences using a scoring scheme in which identical letters score +1 and mismatches score -1. To keep the example simple, ignore spaces and don't allow gaps in the alignment. Although only extension to the right is shown, it also occurs to the left of the seed. Here are the two sentences:
The quick brown fox jumps over the lazy dog. The quiet brown cat purrs when she sees him.
Assume the seed is the capital T, and you're extending it to the right. You can extend the alignment without incident until you get to the first mismatch:
The quic The quie
At this point, a decision must be made about whether to continue the alignment or stop. Looking ahead, it's clear that more letters can be aligned, so it would be foolish to stop now. The ends of the sentences, however, aren't at all similar, so we should stop if there are too many mismatches. To do this, we create a variable, X, that represents the recent alignment history. Specifically, X represents how much the score is allowed to drop off since the last maximum. Let's set X to 5 and see what happens. We'll keep track of the sum score and the drop off score as we go. Figure 5-6 shows the graphical interpretation.
The quick brown fox jump The quiet brown cat purr 123 45654 56789 876 5654 <- score 000 00012 10000 123 4345 <- drop off score
Figure 5-6. Attenuating extension with X
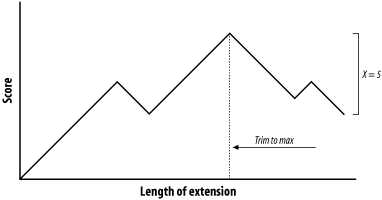
The maximum score for this alignment is 9, and the extension is terminated when the score drops to 4. After terminating, the alignment is trimmed back to the maximum score. If you set X to 1 or 2, the best alignment has a score of 6. If you set X to 3, you can retrieve the longer alignment and save a little computation. A very large value of X doesn't increase the score and requires more computation. So what is the proper value for X? It's generally a good idea to use a large value, which reduces the risk of premature termination and is a better way to increase speed than with the seeding parameters. However, W, I, and 2-hit are better for controlling speed than X.
The gapless extension algorithm just demonstrated is similar to what was used in the original version of BLAST. The algorithms in the current versions of BLAST allow gaps and are related to the dynamic programming techniques described in Chapter 3. Therefore, X not only depends on substitution scores, but also gap initiation and extension costs.
5.2.2.1 Implementation details
Extension in BLASTN is a little different from BLASTP and the other protein-based programs. The reason for this has to do with how nucleotide sequences are stored in BLAST databases. Because there are only four common nucleotide symbols, nucleotide databases can be stored in a highly compressed state with only two bits per nucleotide. What happens if the sequence contains an N or other ambiguous nucleotide? A random canonical nucleotide is substituted. For example, an N can be randomly changed to A, C, G, or T and a Y changed to a C or T. It is possible, especially with long stretches of ambiguous nucleotides, that the two-bit approximation terminates extension prematurely.
NCBI-BLAST and WU-BLAST take very different approaches to the gapped extension procedure. NCBI-BLAST has three values for X (parameters -X -y -Z) and WU-BLAST has two (parameters -X and -gapX). Some differences, such as the presence of a floating bandwidth (NCBI) rather than a fixed bandwidth, are interesting from an academic viewpoint but less so from a user's perspective. What is important is that altering the extension parameters from their defaults is generally not an effective way to increase the speed or sensitivity of a BLAST search. You might consider adjusting the parameters in two situations:
If you align sequences that are nearly identical and you want to prevent the extensions from straying into a nonidentical sequence, you can set the various X values very low.
If you try to align very distant sequences and have already adjusted W, T, and the scoring matrix to allow additional sensitivity, it makes sense to also increase the various X values. That said, when searching for remote homologies, it is often better to employ an algorithm with a position-specific scoring matrix, such as PSI-BLAST or HMMER.
Since gapped extension also depends on the gap initiation and extension costs, you should note that these parameters are interpreted differently in NCBI-BLAST and WU-BLAST. In NCBI-BLAST, the total cost for a gap is the gap opening cost (-G) plus the gap extension cost (-E) times the length of the gap. In WU-BLAST, the total cost of a gap is the cost of the first gap character (-Q) plus all remaining gap characters (-R). The NCBI parameters -G 1 -E 1 are identical to -Q 2 -R 1 in WU-BLAST.
5.2.3 Evaluation
Once seeds are extended in both directions to create alignments, the alignments are evaluated to determine if they are statistically significant (Chapter 4). Those that are significant are termed HSPs. At the simplest level, evaluating alignments is easy; just use a score threshold, S, to sort alignments into low and high scoring. Because S and E are directly related through the Karlin-Altschul equation, a score threshold is synonymous with a statistical threshold. In practice, evaluating alignments isn't as simple, which is due to complications that result from multiple HSPs.
Consider the alignment between a eukaryotic protein and its genomic source. Because most coding regions are broken up by introns, an alignment between the protein and the DNA is expected to produce several HSPs, one for each exon. In assessing the statistical significance of the protein-DNA match, should each exon alignment be forced to stand on its own against the statistical threshold, or does it make more sense to combine the scores of the various exons? The latter is generally more appropriate, especially if some exons are short and may be thrown out if not aided in some way. However, determining the significance of multiple HSPs isn't as simple as summing all the alignment scores because many alignments are expected to be extensions from fortuitous word hits and not all groups of HSPs make sense.
An alignment threshold is an effective way to remove many random, low-scoring alignments (Figure 5-7). However, if the threshold is set too high, (Figure 5-7c), it may also remove real alignments. This alignment threshold is based on score and therefore doesn't consider the size of the database. There are, of course, E-value and P-value interpretations, if you consider the size of individual sequences or a constant theoretical search space.
Figure 5-7. Increasing alignment thresholds remove low scoring alignments

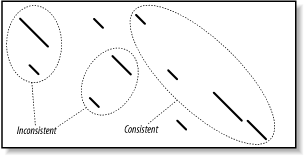
Qualitatively, the relationship between HSPs should resemble the relationship between ungapped alignments. That is, the lines in the graph should start from the upper left and continue to the lower right, the lines shouldn't overlap, and there should be a penalty for unaligned sequence. Groups of HSPs that behave this way are considered consistent. Figure 5-8 shows consistent and inconsistent HSPs. From a biological perspective, you expect the 5´ end of a coding sequence to match the N-terminus of a protein and the 3´ end to match the C-terminus?not vice versa.
Figure 5-8. Consistent and inconsistent alignment groups
The algorithm for defining groups of consistent HSPs compares the coordinates of all HSPs to determine if there are overlaps (a little overlap is actually allowed to account for extensions that may have strayed too far). This computation is quadratic in the number of HSPs and therefore can be costly if there are many HSPs (e.g., when the sequences are long, and the alignment threshold is low).
Once HSPs are organized into consistent groups, they can be evaluated with a final threshold based on the entire search space and corresponding to the value of E set for the search. You can read more about this topic in Chapter 4. BLAST reports any alignment or group of alignments that meets the E requirement.
5.2.3.1 Implementation details
This chapter initially described BLAST as having three phases: seeding, extension, and evaluation. In reality, BLAST isn't so straightforward. There are two rounds of extension and evaluation: ungapped and gapped. Gapped extension and evaluation are triggered only if ungapped alignments surpass the ungapped thresholds. In other words, to find a gapped alignment, you must first find a reasonable ungapped alignment.
In NCBI-BLAST, the command line parameter -e sets the final threshold. The value for the alignment threshold is set by the software and isn't a user-definable parameter. You can find the value for E in the footer. For example, if E is set with -e 1e-10, E is reported as follows:
Number of sequences better than 1.0e-10: 4
The value for the alignment threshold and its gapped equivalent are displayed respectively as S1 and S2 with the raw score listed first. Note that the ungapped threshold is quite a bit lower than the gapped threshold.
S1: 41 (21.7 bits) S2: 158 (65.5 bits)
In WU-BLAST, the E or S parameters specify the final threshold (if both are specified, the most stringent one is used). The command-line parameters S2 and its gapped counterpart gapS2 specify the alignment threshold. WU-BLAST includes E-value versions of the alignment threshold based on a constant search space. They may be set via E2 and gapE2. The values for these parameters are shown in the footer. In this example, the alignment threshold (S2) has an ungapped threshold of 33 and a gapped threshold of 36 (one line below).
Query
Frame MatID Length Eff.Length E S W T X E2 S2
+0 0 235 235 10. 65 3 12 22 0.20 33
32 0.21 36
While gapped alignments are useful from a biological perspective, they pose a small problem to Karlin-Altschul statistics because there is no known way to calculate lambda with arbitrary gap penalties. However, lambda can be estimated by observing the properties of random alignments in a given scoring scheme. Both NCBI-BLAST and WU-BLAST have internal tables that contain Karlin-Altschul parameters for common matrices and gap penalties. If you try to use an unsupported scoring scheme in NCBI-BLAST, the program will terminate and list the possible gap penalties. Unsupported scoring schemes in WU-BLAST revert to ungapped parameters, but a warning is issued that informs you that you can provide your own values for lambda, k, and H on the command line.
NCBI's version of BLASTN doesn't contain gapped values for lambda; lambda is always calculated directly from the match/mismatch scores. Because of this, equivalent alignments may have much higher bit scores (and lower E-values) in NCBI-BLAST than WU-BLAST, even if their match/mismatch scores are identical.







