11.3 Sequence Databases
The sequences in BLAST databases come from sequence databases. But what are sequence databases and where do you get them? The answers to these simple questions are surprisingly complex. Sequence databases come in many shapes and sizes. Some are just collections of raw sequence data from genome sequencing projects, while others contain comprehensive information about the origin and function of the sequences. Unfortunately, there isn't a one-stop shopping place to get all the information you may want, but there is one particular service worth mentioning above all others: the International Nucleotide Sequence Database.
11.3.1 International Nucleotide Sequence Database
Probably the most important molecular biology resource is the public sequence database maintained by the International Nucleotide Sequence Database (INSD). It is composed of three parties: the DNA Data Bank of Japan (DDBJ, http://www.ddbj.nig.ac.jp), the European Molecular Biology Laboratory, (EMBL, http://www.embl.org), and GenBank from the National Center for Biotechnology Information (NCBI, http://ncbi.nlm.nih.gov/GenBank). This consortium collaborates to form the largest public repository for DNA and protein sequences in the world. Because it is such an important resource, this chapter spends some time exploring it.
11.3.2 Database Growth
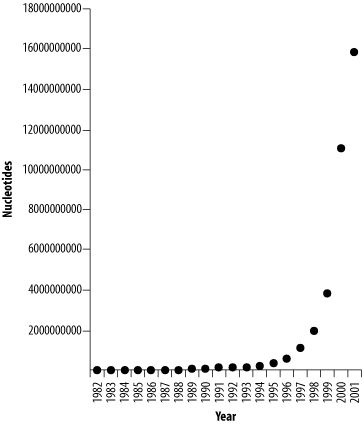
The amount of publicly available sequence has been growing geometrically, doubling approximately every 14 months (see Figure 11-2). Fortunately, computer technology has also kept pace. While it seems scary that GenBank is currently approaching 100 GB and will be half a terabyte in a few years, it's nice to know that this isn't going to be a problem. Not every database grows so fast, though. Organism-specific databases such as the Saccharomyces Genome Database, WormBase, and FlyBase are growing at a more moderate pace, principally because the sequence of their genomes is complete. But many new genome projects are just getting started, and they will probably grow very quickly.
Figure 11-2. Growth of DDBJ/EMBL/GenBank
11.3.3 Flat Files
Sequence databases usually offer their data in several different formats. The FASTA format is universally accepted for operating on sequences, but many sequence databases record a lot more data than just the sequence. Such extra information is commonly presented in a human-readable format called a flat file. The INSD uses two kinds of flat files. The DDBJ and GenBank flat file formats are identical, while the EMBL format is slightly different. The following DDBJ/GenBank record corresponds to a fragment of the Hoxa-11 gene from the coelacanth (the ancient fish on the cover of the book):
LOCUS AF287139 606 bp DNA linear VRT 10-DEC-2000
DEFINITION Latimeria chalumnae Hoxa-11 gene, partial cds.
ACCESSION AF287139
VERSION AF287139.1 GI:11611818
KEYWORDS .
SOURCE Latimeria chalumnae.
ORGANISM Latimeria chalumnae
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
Coelacanthiformes; Coelacanthidae; Latimeria.
REFERENCE 1 (bases 1 to 606)
AUTHORS Chiu,C.H., Nonaka,D., Xue,L., Amemiya,C.T. and Wagner,G.P.
TITLE Evolution of Hoxa-11 in lineages phylogenetically positioned along
the fin-limb transition
JOURNAL Mol. Phylogenet. Evol. 17 (2), 305-316 (2000)
MEDLINE 20538275
PUBMED 11083943
REFERENCE 2 (bases 1 to 606)
AUTHORS Chiu,C.-H. and Wagner,G.P.
TITLE Direct Submission
JOURNAL Submitted (14-JUL-2000) Ecology and Evolutionary Biology, Yale
University, 165 Prospect St., New Haven, CT 06520-8106, USA
FEATURES Location/Qualifiers
source 1..606
/organism="Latimeria chalumnae"
/db_xref="taxon:7897"
CDS <1..>606
/codon_start=1
/product="Hoxa-11"
/protein_id="AAG39070.1"
/db_xref="GI:11611819"
/translation="YLPSCTYYVSGPDFSSLPSFLPQTPSSRPMTYSYSSNLPQVQPV
REVTFRDYAIDTSNKWHPRSNLPHCYSTEEILHRDCLATTTASSIGEIFGKGNANVYH
PGSSTSSNFYNTVGRNGVLPQAFDQFFETAYGTTENHSSDYSADKNSDKIPSAATSRS
ETCRETDEKERREESSSPESSSGNNEEKSSSSSGQRTRKKRC"
BASE COUNT 173 a 169 c 129 g 135 t
ORIGIN
1 tacttgccaa gttgcaccta ctacgtttcg ggtcccgatt tctccagcct cccttctttt
61 ttgccccaga ccccgtcttc tcgccccatg acatactcct attcgtctaa tctaccccaa
121 gttcaacctg tgagagaagt taccttcagg gactatgcca ttgatacatc caataaatgg
181 catcccagaa gcaatttacc ccattgctac tcaacagagg agattctgca cagggactgc
241 ctagcaacca ccaccgcttc aagcatagga gaaatctttg ggaaaggcaa cgctaacgtc
301 taccatcctg gctccagcac ctcttctaat ttctataaca cagtgggtag aaacggggtc
361 ctaccgcaag cctttgacca gtttttcgag acggcttatg gcacaacaga aaaccactct
421 tctgactact ctgcagacaa gaattccgac aaaatacctt cggcagcaac ttcaaggtcg
481 gagacttgca gggagacaga cgagaaggag agacgggaag aaagcagtag cccagagtct
541 tcttccggca acaatgagga gaaatcaagc agttccagtg gtcaacgtac aaggaagaag
601 aggtgc
//
The next example is the same record in the slightly different EMBL format. Most of the data is identical between the two formats, but there are a few important differences. The VERSION field of the DDBJ/GenBank record includes a GI number (discussed below) that isn't in the EMBL record. The EMBL record contains both a creation date and a modification date, while the DDBJ/GenBank record contains only a modification date.
ID AF287139 standard; DNA; VRT; 606 BP.
XX
AC AF287139;
XX
SV AF287139.1
XX
DT 11-DEC-2000 (Rel. 66, Created)
DT 11-DEC-2000 (Rel. 66, Last updated, Version 1)
XX
DE Latimeria chalumnae Hoxa-11 gene, partial cds.
XX
KW .
XX
OS Latimeria chalumnae (coelacanth)
OC Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi;
OC Coelacanthiformes; Coelacanthidae; Latimeria.
XX
RN [1]
RP 1-606
RX PUBMED; 11083943.
RA Chiu, Ch, Nonaka D., Xue L., Amemiya C.T., Wagner G.P.;
RT "Evolution of Hoxa-11 in Lineages Phylogenetically Positioned along the
RT Fin-Limb Transition";
RL Mol. Phylogenet. Evol. 17(2):305-316(2000).
XX
RN [2]
RP 1-606
RA Chiu C.-H., Wagner G.P.;
RT ;
RL Submitted (14-JUL-2000) to the RL Ecology and Evolutionary Biology, Yale
University, 165 Prospect St., New
RL Haven, CT 06520-8106, USA
XX
DR SPTREMBL; Q9DDT9; Q9DDT9.
XX
FH Key Location/Qualifiers
FH
FT source 1..606
FT /db_xref="taxon:7897"
FT /organism="Latimeria chalumnae"
FT CDS <1..>606
FT /codon_start=1
FT /db_xref="SPTREMBL:Q9DDT9"
FT /product="Hoxa-11"
FT /protein_id="AAG39070.1"
FT /translation="YLPSCTYYVSGPDFSSLPSFLPQTPSSRPMTYSYSSNLPQVQPVR
FT EVTFRDYAIDTSNKWHPRSNLPHCYSTEEILHRDCLATTTASSIGEIFGKGNANVYHPG
FT SSTSSNFYNTVGRNGVLPQAFDQFFETAYGTTENHSSDYSADKNSDKIPSAATSRSETC
FT RETDEKERREESSSPESSSGNNEEKSSSSSGQRTRKKRC"
XX
SQ Sequence 606 BP; 173 A; 169 C; 129 G; 135 T; 0 other;
tacttgccaa gttgcaccta ctacgtttcg ggtcccgatt tctccagcct cccttctttt 60
ttgccccaga ccccgtcttc tcgccccatg acatactcct attcgtctaa tctaccccaa 120
gttcaacctg tgagagaagt taccttcagg gactatgcca ttgatacatc caataaatgg 180
catcccagaa gcaatttacc ccattgctac tcaacagagg agattctgca cagggactgc 240
ctagcaacca ccaccgcttc aagcatagga gaaatctttg ggaaaggcaa cgctaacgtc 300
taccatcctg gctccagcac ctcttctaat ttctataaca cagtgggtag aaacggggtc 360
ctaccgcaag cctttgacca gtttttcgag acggcttatg gcacaacaga aaaccactct 420
tctgactact ctgcagacaa gaattccgac aaaatacctt cggcagcaac ttcaaggtcg 480
gagacttgca gggagacaga cgagaaggag agacgggaag aaagcagtag cccagagtct 540
tcttccggca acaatgagga gaaatcaagc agttccagtg gtcaacgtac aaggaagaag 600
aggtgc 606
//
Note that the sequence data is only one part of the record; there's a lot of other useful information in here including the organism, the taxonomic classification, the authors, a reference to the scientific literature, and a feature table indicating the translation of the DNA. This is great stuff, and INSD is full of these kinds of records. But there is a downside to using the public databases. They're a bit like public parks: huge, beautiful, inexpensive to use, and valuable, but there's always someone who doesn't pick up their trash. Some sequences are erroneous, and the ancillary information is sometimes wrong and misleading. But overall, the databases are high-quality resources, and you should take a moment to applaud the scientists who contribute their sequences to the INSD, as well as the administrators and curators at DDBJ/EMBL/GenBank who do an outstanding job. Now let's take a closer look at some parts of the sequence record.
11.3.3.1 ACCESSION, LOCUS, VERSION, and GI
One of the most important parts of any sequence record is its database identifier, which is often called its accession number. (Although it's called a number, it may be a mixture of letters, numbers, and other symbols, but not spaces.) This tag uniquely identifies the sequence in a database. There isn't necessarily a one-to-one correspondence between sequences and tags because sequences are sometimes known by multiple unique names. The DDBJ/GenBank ACCESSION (or AC in EMBL) is the primary name for a sequence record. Another unique name is the LOCUS (or ID in EMBL). The locus is supposed to be a "short mnemonic name for the entry, chosen to suggest the sequence's definition." For example, "HSMG01" is the locus name for the database entry containing Homo sapiens myoglobin exon 1. Over time, like the names of celestial objects, locus names have become less descriptive and are often just duplicates of the accession numbers.
Sequence records can also change over time. This often happens when the record is edited to correct a sequence error. The accession number and locus don't change, but the version number is increased (VERSION in DDBJ/GenBank and SV in EBML). In this way, an ACCESSION.VERSION points to a particular record at a particular time. It's a good idea to always refer to sequences in this way and not by ACCESSION alone or by LOCUS or ID.
DDBJ/GenBank records include an additional token called the GI number, which is a numeric identifier that points to a particular ACCESSION.VERSION. The GI number is especially important because NCBI-BLAST relies on it as an additional mechanism for indexing BLAST databases. This topic was covered in Section 11.2.3.
11.3.3.2 DEFINITION, KEYWORDS, and SOURCE
The DEFINITION is a concise description of the origin and function of a sequence, and is typically what you find a FASTA description. The text is structured, meaning that there are rules that define how it is produced. However, it doesn't use a controlled vocabulary, which means you can't be sure which words will or won't appear.
KEYWORDS are a historical relic like the locus name and aren't used in modern sequence records. Avoid the temptation to believe that keywords are meaningful.
The common name for an organism is often found in the SOURCE, or in parentheses after the OS in EMBL format. The scientific name is on the ORGANISM line (OS in EMBL) and the complete taxonomic classification is given on the following lines (OC in EMBL). The complete taxonomy may be abbreviated if it's especially long.
11.3.3.3 FEATURES
The FEATURES (FT in EMBL) list specific regions of importance on the sequence such as genes or repetitive elements. The general syntax of features is fairly simple; each has a key and location, and optional qualifiers. The key tells what kind of feature it is (e.g., a gene), the location (e.g., from nucleotide 100 to nucleotide 200), and the qualifiers include additional information, such as specific names, database cross references, and experimental notes. A detailed discussion of the feature table is beyond the scope of this book. See http://www.ncbi.nih.gov/projects/collab/FT for more information.
11.3.4 Other Common Databases
INSD is just one of many important databases. Some other favorites are listed in Table 11-2.
|
Database |
Description | ||
|---|---|---|---|
|
RefSeq |
RefSeq provides reference sequences that represent the highest quality information about a particular sequence. Each record may be constructed from several INSD records, which makes the database nonredundant. All RefSeq accession numbers are preceded by two letters and an underscore, for example XP_102310. Some types of RefSeq records have been inspected manually by curators, and they are the highest quality records (indicated below). | ||
|
Prefix |
Molecule |
Description | |
|
NC_ |
Genomic |
Curated complete genomic molecules including genomes, chromosomes, organelles, and plasmids. | |
|
NG_ |
Genomic |
Curated incomplete genomic region; primarily supplied for Homo sapiens and Mus musculus to support the NCBI Genome Annotation pipeline. | |
|
NM_ |
mRNA |
Curated mRNAs. | |
|
NR_ |
RNA |
Curated noncoding transcripts including structural RNAs, transcribed pseudogenes, and others. | |
|
NP_ |
Protein |
Curated proteins. | |
|
NT_ |
Genomic |
Intermediate genomic assemblies of BAC sequence data. | |
|
NW_ |
Genomic |
Intermediate genomic assemblies of Whole Genome Shotgun sequence data. | |
|
XM_ |
mRNA |
Homo sapiens model mRNA provided by the Genome Annotation process; sequence corresponds to the genomic contig. | |
|
XR_ |
RNA |
Homo sapiens model noncoding transcripts provided by the Genome Annotation process; sequence corresponds to the genomic contig. | |
|
http://www.ncbi.nlm.nih.gov/LocusLink/refseq.html | |||
|
Pfam |
Pfam is a collection of multiple sequence alignments and hidden Markov models (HMMs) for many common protein domains and families. If you are interested in a particular family, such as globins, or a particular domain, such as WD-40, this is a great resource. HMMs are probabilistic models that describe how whole domains evolve, which is quite different from a scoring matrix employed by BLAST that treats each amino acid of a protein independently. http://pfam.wustl.edu,http://www.sanger.ac.uk/pfam | ||
|
SWISS-PROT |
The SWISS-PROT Protein Knowledgebase is a curated protein sequence database that provides a high level of annotation (such as the description of protein function, domains structure, post-translational modifications, variants, etc.), a minimal level of redundancy, and high level of integration with other databases. http://www.ebi.ac.uk/swissprot | ||
|
TrEMBL |
The TrEMBL database contains translations of all coding sequences (CDS) present in the INSD, which aren't yet integrated into SWISS-PROT. TrEMBL is split into two main sections: SP-TrEMBL contains entries expected to be included in SWISS-PROT, and REM-TrEMBL contains those that aren't expected to be included. http://www.ebi.ac.uk/tremble | ||
|
UniGene |
UniGene is an experimental system for automatically partitioning GenBank sequences into a nonredundant set of gene-oriented clusters. Each UniGene cluster contains sequences that represent a unique gene, as well as related information such as the tissue types in which the gene has been expressed and the map location. UniGene sets are available for most genomes with a lot of EST sequences. http://www.ncbi.nlm.nih.gov/UniGene | ||
|
MGC |
The goal of the Mammalian Gene Collection (MGC) is to provide a complete set of full-length (open reading frame) sequences and cDNA clones of expressed mammalian genes. The current focus is limited to human and mouse. http://mgc.nci.nih.gov | ||
|
SGD |
SGD is a scientific database of the molecular biology and genetics of the yeast Saccharomyces cerevisiae, which is commonly known as baker's or budding yeast. S. cerevisiae was the first eukaryotic genome sequenced. http://genome-www.stanford.edu/Saccharomyces | ||
|
WormBase |
WormBase is a comprehensive database dedicated to the biology and genome of the nematode Caenorhabditis elegans. C. elegans was the first multicellular organism to have its genome sequenced. http://www.wormbase.org | ||
|
FlyBase |
FlyBase is a comprehensive database for information on the genetics and molecular biology of Drosophila. It includes data from the Drosophila Genome Projects and data curated from the literature. FlyBase is a joint project with the Berkeley Drosophila Genome Project. http://www.flybase.org | ||
|
TAIR |
The Arabidopsis Information Resource (TAIR) provides a comprehensive resource for the scientific community working with Arabidopsis thaliana, a widely used model plant. http://www.arabidopsis.org | ||







