1.4 Alternate Alignment Views
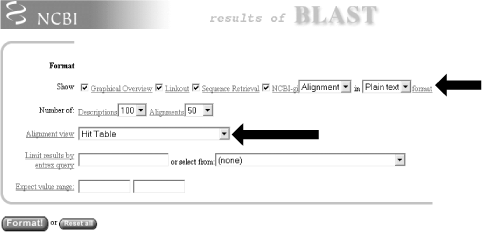
The default Pairwise view shown in Figure 1-10 is the classic BLAST output style, but other options are available for other purposes. These options, described in the NCBI reference section and in Appendix A, include pairwise, query-anchored with identities, query-anchored without identities, flat query-anchored with identities, flat query-anchored without identities, and Hit Table. The most friendly option for text parsers is the Hit Table, which is viewed in plaintext format. This displays all the results in a tab-delimited table, which can be parsed easily. You can select this at the top of the page by changing "Format" to "Plain text" and "Alignment view" to "Hit Table" (Figure 1-12).
Figure 1-12. Changing format options
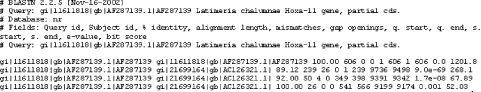
The Hit Table alignment view is shown in Figure 1-13. The first five lines start with # and are comments about the BLAST program, the query, and the database, followed by a description of the reported fields. The lines after the comments are the alignments in table format. The Hit Table contains all the necessary data to judge a hit without displaying the actual sequence being aligned.
Figure 1-13. Hit Table alignment
The other available alignment options allow a multiple sequence alignment view of the BLAST hits. One of these multiple alignment options, query-anchored with identities, is shown in Figure 1-14. In this view, the full sequence of the query is shown on the top line with a unique identifier (1_18852, in this case). Subsequently, each line shows the alignment for one database hit. Identical residues are represented with a dot (.), while nucleotide differences are shown explicitly. This alignment option is useful for quickly identifying changes common to a group of sequences. For example, you can see from the part of the alignment shown in Figure 1-14 that the bottom four sequences (6754225, 664837, 664835, and 664831) have common shared differences. A deeper look into these sequences reveals that they are actually different database entries for the same mouse Hoxa11 gene, which is homologous to the coelacanth Hoxa11 gene.
Figure 1-14. Query-anchored with identities view

The other multiple sequence alignment views are similar to this one, but differ on whether or not they show identical residues (with or without identities) and whether the gaps are displayed in the query sequence or in the subjects (flat or not). You'll find a detailed explanation of these alignment options in Appendix A.








